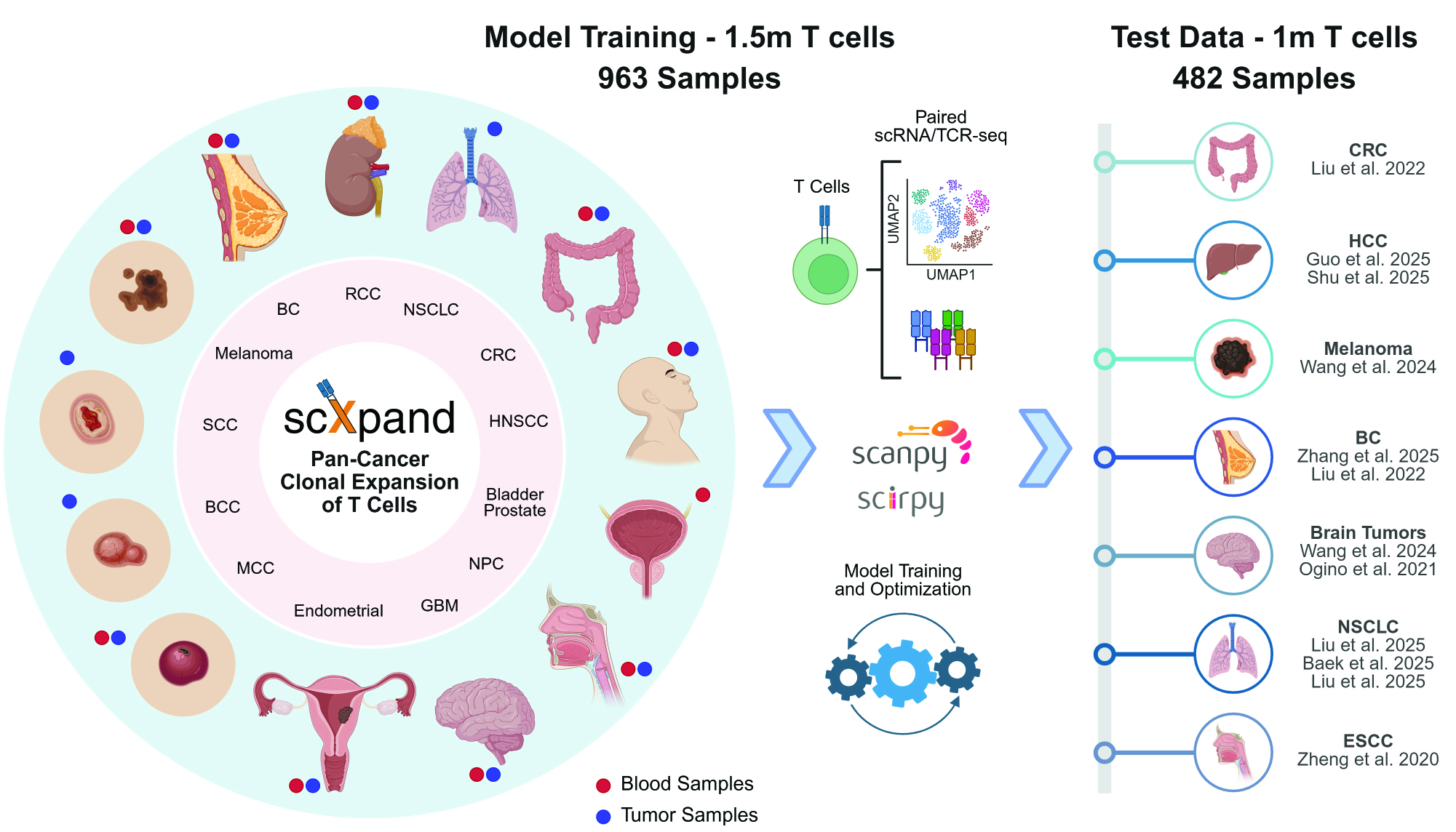
scXpand: Pan-cancer Detection of T-cell Clonal Expansion#
scXpand is a framework for predicting T-cell clonal expansion from single-cell RNA sequencing data without paired TCR sequencing. It provides multiple methods for training and inference.
GitHub Repository: yizhak-lab-ccg/scXpand
Preprint: https://www.biorxiv.org/content/10.1101/2025.09.14.676069v1
Features#
Multiple Model Architectures: Autoencoder, MLP, LightGBM, Logistic Regression, and SVM
Scalable Processing: Handles millions of cells with memory-efficient data streaming
Hyperparameter Optimization: Built-in hyperparameter search for model tuning
Installation#
For complete installation instructions including prerequisites, package installation, and development setup, please see our Installation Guide guide.
Quick Start#
import scxpand
# Make sure that "your_data.h5ad" includes only T cells for the results to be meaningful
# scXpand requires raw UMI counts. Normalized or log-transformed data is not supported
# Ensure that "your_data.var_names" are provided as Ensembl IDs (as the pre-trained models were trained using this gene representation)
# Please refer to our documentation for more information
# List available pre-trained models
scxpand.list_pretrained_models()
# Run inference with automatic model download
results = scxpand.run_inference(
model_name="pan_cancer_autoencoder", # default model
data_path="your_data.h5ad"
)
# Access predictions
predictions = results.predictions
if results.has_metrics:
print(f"AUROC: {results.get_auroc():.3f}")
# Or use the command line
# scxpand inference --data_path your_data.h5ad --model_name pan_cancer_autoencoder
See our User Guide for comprehensive usage instructions.
Tutorials#
We provide a variety of tutorials to help you get started with scXpand:
Predicting T Cell Expansion from scRNA-seq Data - download example scRNA-seq dataset (with no paired TCR-seq) and apply scXpand models for T cell expansion prediction using a breast cancer dataset example.
Preparing Training Data from Paired scRNA/TCR-seq - Complete pipeline for preparing labeled data (expansion status and tissue type) from paired scRNA/TCR-seq data, including quality control, MAGIC imputation, automatic cutoff determination for cell type classification, and expansion labeling.
Model Inference and Evaluation Pipeline - Load trained models, run inference on labeled data, and evaluate performance using ROC curves and AUROC metrics across different tissue types and labels.
Autoencoder Embedding Visualization - Generate and visualize latent representations from autoencoder models, coloring plots by expansion status and tissue type for biological insights.
Support and Contact#
This project was created in favor of the scientific community worldwide, with a special dedication to the cancer research community. We hope you’ll find this repository helpful, and we warmly welcome any requests or suggestions - please don’t hesitate to reach out!
Citation#
If you use scXpand in your research, please cite our paper:
Shorer, O., Amit, R., and Yizhak, K. (2025). scXpand: Pan-cancer detection of T-cell clonal expansion from single-cell RNA sequencing without paired single-cell TCR sequencing. Preprint at bioRxiv, https://doi.org/10.1101/2025.09.14.676069.
BibTeX
@article{shorer2025scxpand,
title={scXpand: Pan-cancer detection of T-cell clonal expansion from single-cell RNA sequencing without paired single-cell TCR sequencing},
author={Shorer, Ofir and Amit, Ron and Yizhak, Keren},
year={2025},
journal={bioRxiv},
doi={https://doi.org/10.1101/2025.09.14.676069}
}