Predicting T Cell Expansion from scRNA-seq Data#
This tutorial demonstrates how to prepare scRNA-seq data and apply scXpand models for T cell expansion prediction.
Example Dataset#
We use a publicly available scRNA-seq dataset of breast cancer patients from:
Study: Tietscher et al. 2023 (E-MTAB-10607)
Source: https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-10607/
Data type: scRNA-seq only (no paired scTCR-seq)
Cancer type: Breast cancer
Tissue: Tumor samples
Tutorial Structure#
Data Loading and Initial Processing
Quality Control and Filtering
Data Preparation for scXpand
Model Application and Inference
Robustness Testing
Data Loading and Initial Processing#
import sys
from pathlib import Path
import anndata as ad
import matplotlib.pyplot as plt
import numpy as np
import pandas as pd
import scanpy as sc
import seaborn as sns
from scipy import sparse
# Set matplotlib backend for Jupyter notebooks
%matplotlib inline
# Plotting settings
plt.rcParams["font.sans-serif"] = ["Arial"]
plt.rcParams["axes.axisbelow"] = True
sns.set_style("whitegrid")
# Setup project paths
project_root = Path.cwd().parent.parent
print(f"Project root: {project_root}")
sys.path.insert(0, str(project_root))
Project root: c:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand
Discover and Validate Data Files#
# Setup data paths
demo_path = project_root / "data" / "demo"
source_path = demo_path / "E-MTAB-10607_BC"
# Please make sure to download the data from https://www.ebi.ac.uk/biostudies/arrayexpress/studies/E-MTAB-10607/ and place it in the source_path directory
# Collect matrix and metadata files
mtx_files = sorted(source_path.glob("*_matrix.txt"))
metadata_files = sorted(source_path.glob("*_metadata.txt"))
# Extract sample IDs from matrix file prefixes
sample_ids = [file.stem.split("_")[0] for file in mtx_files]
# Validate file consistency: sample_ids should match the metadata_files and mtx_files file names
assert len(sample_ids) == len(metadata_files) == len(mtx_files)
assert all(
sample_id in metadata_files[i].stem for i, sample_id in enumerate(sample_ids)
)
assert all(sample_id in mtx_files[i].stem for i, sample_id in enumerate(sample_ids))
n_samples = len(mtx_files)
print(f"Found {n_samples} samples to process")
Found 14 samples to process
Load and Concatenate Sample Data#
def load_sample_data(sample_id: str, mtx_file: Path, metadata_file: Path) -> ad.AnnData:
"""Load and process a single sample's data."""
# Load expression matrix as AnnData and transpose
sample_data = sc.read_text(mtx_file).transpose()
# Create clean cell names (concatenate sample_id with cell_id)
sample_data.obs_names = (
sample_id + "_" + sample_data.obs_names.to_series().str.split(".").str[0]
)
# Convert to sparse matrix for memory efficiency
sample_data.X = sparse.csr_matrix(sample_data.X)
# Load and merge metadata
metadata = pd.read_csv(metadata_file, sep="\t", index_col=0)
for col in metadata.columns:
sample_data.obs[col] = metadata[col]
return sample_data
# Load samples with progress tracking
print("Loading all samples...")
adata_list = []
for sample_idx in range(n_samples):
print(f"Loading sample {sample_idx + 1} of {n_samples}: {sample_ids[sample_idx]}")
sample_adata = load_sample_data(
sample_id=sample_ids[sample_idx],
mtx_file=mtx_files[sample_idx],
metadata_file=metadata_files[sample_idx],
)
adata_list.append(sample_adata)
# Concatenate all samples
adata = ad.concat(adata_list)
del adata_list # Free memory
Loading all samples...
Loading sample 1 of 14: TBB011
Loading sample 2 of 14: TBB035
Loading sample 3 of 14: TBB075
Loading sample 4 of 14: TBB102
Loading sample 5 of 14: TBB111
Loading sample 6 of 14: TBB129
Loading sample 7 of 14: TBB165
Loading sample 8 of 14: TBB171
Loading sample 9 of 14: TBB184
Loading sample 10 of 14: TBB212
Loading sample 11 of 14: TBB214
Loading sample 12 of 14: TBB226
Loading sample 13 of 14: TBB330
Loading sample 14 of 14: TBB338
print(f"Combined dataset shape: {adata.shape}")
print(f"Data type: {type(adata.X)}")
Combined dataset shape: (159498, 21959)
Data type: <class 'scipy.sparse._csr.csr_matrix'>
Quality Control and Filtering#
Mitochondrial Gene Analysis#
# Display all mitochondrial genes in dataset (37 genes total)
mt_genes_in_data = adata.var[adata.var_names.str.startswith("MT-")].index
print(f"Found {len(mt_genes_in_data)} mitochondrial genes in dataset:")
print(mt_genes_in_data.tolist())
Found 37 mitochondrial genes in dataset:
['MT-TF', 'MT-RNR1', 'MT-TV', 'MT-RNR2', 'MT-TL1', 'MT-ND1', 'MT-TI', 'MT-TQ', 'MT-TM', 'MT-ND2', 'MT-TW', 'MT-TA', 'MT-TN', 'MT-TC', 'MT-TY', 'MT-CO1', 'MT-TS1', 'MT-TD', 'MT-CO2', 'MT-TK', 'MT-ATP8', 'MT-ATP6', 'MT-CO3', 'MT-TG', 'MT-ND3', 'MT-TR', 'MT-ND4L', 'MT-ND4', 'MT-TH', 'MT-TS2', 'MT-TL2', 'MT-ND5', 'MT-ND6', 'MT-TE', 'MT-CYB', 'MT-TT', 'MT-TP']
# We will use only the 13 protein-coding mitochondrial genes for QC
# (to be consistent with other datasets having just these genes)
mt_genes = [
"MT-ND1",
"MT-ND2",
"MT-CO1",
"MT-CO2",
"MT-ATP8",
"MT-ATP6",
"MT-CO3",
"MT-ND3",
"MT-ND4L",
"MT-ND4",
"MT-ND5",
"MT-ND6",
"MT-CYB",
]
# Mark protein-coding mitochondrial genes for QC
adata.var["mt"] = adata.var_names.isin(mt_genes)
print(f"Using {adata.var['mt'].sum()} protein-coding MT genes for QC")
# Calculate QC metrics for each cell
sc.pp.calculate_qc_metrics(adata, qc_vars=["mt"], inplace=True)
Using 13 protein-coding MT genes for QC
Quality Control Visualization and Filtering#
# Visualize mitochondrial gene percentage distribution
ax = sc.pl.violin(adata, "pct_counts_mt", jitter=0.1, show=False)
ax.set_title("Mitochondrial gene percentage distribution")
plt.show()
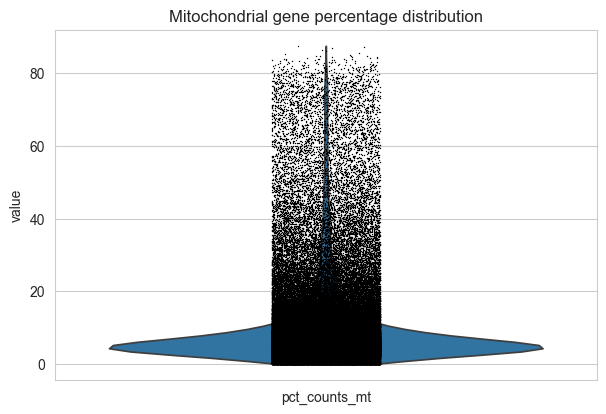
# Apply quality control filters
print(f"Before filtering: {adata.n_obs} cells, {adata.n_vars} genes")
# Filter cells with high mitochondrial content (< 10%)
adata = adata[adata.obs.pct_counts_mt < 10, :].copy()
# Filter low-quality cells and genes
sc.pp.filter_cells(adata, min_genes=200) # Remove cells with < 200 genes
sc.pp.filter_genes(adata, min_cells=3) # Remove genes in < 3 cells
print(f"After filtering: {adata.n_obs} cells, {adata.n_vars} genes")
print(f"Final data shape: {adata.shape}")
Before filtering: 159498 cells, 21959 genes
After filtering: 119704 cells, 19754 genes
Final data shape: (119704, 19754)
Doublet Detection#
Simulates doublets by combining random cell pairs
Computes doublet scores for all cells
Provides automatic threshold detection
Processes samples separately to account for batch effects
# Run Scrublet doublet detection
sc.pp.scrublet(
adata,
expected_doublet_rate=0.05, # Expected doublet rate for the dataset
batch_key="sample", # Process each sample separately
random_state=42, # For reproducibility
)
# Original code used in our dataset generation (replaced with scanpy's implementation due to compatibility issues in non-Windows environments):
# import scrublet as scr
# scrub = scr.Scrublet(adata.X, expected_doublet_rate=0.05)
# adata.obs["doublet_scores"], adata.obs["predicted_doublets"] = scrub.scrub_doublets()
# Plot histogram of doublet scores (optional)
# sc.pl.scrublet_score_distribution(adata)
# Filter out cells with high doublet score using a conservative threshold
doublet_threshold = 0.3
adata = adata[adata.obs["doublet_score"] < doublet_threshold]
# Note: column name is "doublet_score" not "doublet_scores"
print(f"Filtered out cells with doublet scores >= {doublet_threshold}")
print(f"Remaining cells after doublet filtering: {adata.n_obs}")
Filtered out cells with doublet scores >= 0.3
Remaining cells after doublet filtering: 118631
# Verify doublet filtering results
print("Predicted doublet status:")
print(adata.obs["predicted_doublet"].value_counts())
print(f"Max doublet score after filtering: {adata.obs['doublet_score'].max():.3f}")
# filter remaining doublets based on predicted_doublet column:
adata = adata[~adata.obs["predicted_doublet"], :].copy()
print(f"Remaining cells after removing predicted doublets: {adata.n_obs}")
Predicted doublet status:
predicted_doublet
False 118517
True 114
Name: count, dtype: int64
Max doublet score after filtering: 0.299
Remaining cells after removing predicted doublets: 118517
# save raw count matrix:
adata.layers["counts"] = adata.X.copy()
# add some metadata information for our records:
adata.obs["cancer_type"] = "BC"
adata.obs["study"] = "Tietscher et al. 2023 (BC)"
adata.obs["tissue_type"] = "Tumor"
# apply this because of file-writing error in this specific dataset:
adata.obs["excl_doublet"] = adata.obs["excl_doublet"].astype(str)
adata.obs["excl_QC"] = adata.obs["excl_QC"].astype(str)
Data Preparation for scXpand#
T Cell Filtering#
The scXpand model is specifically designed for T cells. We need to filter our dataset
to include only T cells before applying the model.
Note: This step can be done using:
Your own clustering solution
Cell type annotation tools
Imputation methods (e.g., MAGIC)
Pre-existing cell annotations (as in this example)
# Filter for T cells only (removing NK cells) according to cell metadata from authors
# The model input will include only T cells (we kept NKT cells as well)
print(f"Before T cell filtering: {adata.n_obs} cells")
t_cell_mask = ~adata.obs["Tcell_metacluster"].isin(
{"none", "NK", "NK_activated", np.nan}
)
adata = adata[t_cell_mask]
print(f"After T cell filtering: {adata.n_obs} cells remaining")
Before T cell filtering: 118517 cells
After T cell filtering: 26596 cells remaining
Gene ID Conversion#
Important: The scXpand model requires Ensembl gene IDs as gene identifiers.
If your data already has Ensembl IDs: Simply set them as
var_namesand the model will handle missing genes automaticallyIf your data uses gene symbols: Follow the conversion process below
# Load the gene list we want to keep (authors provided genes without ensembl_ids)
# This file can be found in our Figshare publicly available repository: https://doi.org/10.6084/m9.figshare.30067666
gene_table_path = demo_path / "gene_table_for_scXpand.csv.gz"
gene_table = pd.read_csv(gene_table_path, index_col=1)
print(f"Loaded gene table with {len(gene_table)} genes")
print("Gene table columns:", gene_table.columns.tolist())
Loaded gene table with 11950 genes
Gene table columns: ['gene_ids', 'feature_types']
# Filter gene table to include only genes present in the dataset
# Note: If genes were missing, the model would handle this by setting expression to zero
gene_table = gene_table[gene_table.index.to_series().isin(adata.var.index)]
print(f"Gene table after filtering: {len(gene_table)} genes")
print(f"All genes present in dataset: {len(gene_table) == len(adata.var)}")
Gene table after filtering: 11950 genes
All genes present in dataset: False
# Subset adata to include only genes in the gene table
adata = adata[:, gene_table.index].copy()
print(f"Data after gene filtering: {adata.shape}")
# Add gene IDs and feature types to adata.var
adata.var["gene_ids"] = gene_table["gene_ids"]
adata.var["feature_types"] = gene_table["feature_types"]
Data after gene filtering: (26596, 11950)
# Convert gene names to Ensembl IDs (required by scXpand model)
# The model requires Ensembl IDs as var_names (index)
adata.var = adata.var.rename_axis("gene_name").reset_index().set_index("gene_ids")
print("Gene identifiers successfully converted to Ensembl IDs")
print(f"Example gene IDs: {adata.var.index[:5].tolist()}")
Gene identifiers successfully converted to Ensembl IDs
Example gene IDs: ['ENSG00000188976', 'ENSG00000187961', 'ENSG00000187583', 'ENSG00000188290', 'ENSG00000187608']
Save Processed Data (Optional)#
# Save the processed data for future use (optional)
output_path = (
project_root
/ "data"
/ "demo"
/ "processed"
/ "scXpand_count_data_T_cells_E-MTAB-10607.h5ad"
)
adata.write(output_path, compression="gzip")
print(f"Processed data saved to: {output_path}")
print("Data preprocessing completed - ready for model application!")
Processed data saved to: c:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\data\demo\processed\scXpand_count_data_T_cells_E-MTAB-10607.h5ad
Data preprocessing completed - ready for model application!
T Cell Expansion Prediction with Pre-trained Models#
Configure Model and Data#
# Model configuration
model_name = "pan_cancer_logistic" # Choose from available pretrained models
# Data configuration - we'll use the processed data from the demo
adata_path = demo_path / "processed" / "scXpand_count_data_T_cells_E-MTAB-10607.h5ad"
adata_demo = sc.read_h5ad(adata_path) # Load preprocessed data
# Inference parameters
batch_size = 2048
num_workers = 4
save_path = project_root / "results" / "inference_results" / f"{model_name}_inference"
print(f"Selected model: {model_name}")
print(f"Demo data: {adata_demo.n_obs} cells, {adata_demo.n_vars} genes")
print(f"Results will be saved to: {save_path}")
# Create output directory
save_path.mkdir(parents=True, exist_ok=True)
Selected model: pan_cancer_logistic
Demo data: 26596 cells, 11950 genes
Results will be saved to: c:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\results\inference_results\pan_cancer_logistic_inference
Run Inference#
# Run inference on the demo dataset
from scxpand import run_inference
print(f"Running inference with {model_name} model...")
results = run_inference(
adata=adata_demo,
model_name=model_name,
batch_size=batch_size,
num_workers=num_workers,
)
y_pred_prob = results.predictions
print("Inference completed successfully!")
print(f"Example predictions (probabilities): {y_pred_prob[:5]}")
print(f"Prediction shape: {y_pred_prob.shape}")
Running inference with pan_cancer_logistic model...
2025-10-10 11:30:28 [info ] Downloading registry model 'pan_cancer_logistic' to cache directory: c:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\docs\notebooks\.scxpand_cache [scxpand.pretrained.download_manager]
2025-10-10 11:30:29 [info ] Loaded data format from: C:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\docs\notebooks\.scxpand_cache\5be9d6a5c323e8814ceae709733c7003-1.unzip\logistic\data_format.json [scxpand.data_util.data_format]
2025-10-10 11:30:29 [info ] Loading logistic model from C:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\docs\notebooks\.scxpand_cache\5be9d6a5c323e8814ceae709733c7003-1.unzip\logistic [scxpand.util.inference_utils]
2025-10-10 11:30:29 [info ] Inference environment ready: logistic model on cuda [scxpand.util.inference_utils]
2025-10-10 11:30:29 [info ] Running inference... [scxpand.core.prediction]
2025-10-10 11:30:29 [info ] Created eval data loader with batch size: 2048, num_workers: 4 [scxpand.data_util.dataloaders]
2025-10-10 11:30:47 [info ] Inference completed. Generated 26596 predictions. [scxpand.core.prediction]
2025-10-10 11:30:47 [info ] Missing ['expansion'] columns in observation data. Skipping metrics evaluation. [scxpand.core.evaluation]
Inference completed successfully!
Example predictions (probabilities): [0.76589788 0.54469571 0.14568532 0.78571869 0.60704418]
Prediction shape: (26596,)
Robustness Testing#
Gene Order Independence Test#
The scXpand model is designed to be robust to gene ordering. Let’s verify this
by shuffling the genes and confirming identical predictions.
# Shuffle genes to demonstrate gene-order independence
print("Testing gene-order independence...")
shuffled_var = adata_demo.var.sample(frac=1, random_state=42)
results = run_inference(
model_name=model_name,
adata=adata_demo[:, shuffled_var.index],
batch_size=batch_size,
num_workers=num_workers,
)
y_pred_prob_shuffled = results.predictions
print("Inference with shuffled genes completed!")
print(f"Example predictions (probabilities): {y_pred_prob_shuffled[:5]}")
# Verify predictions are identical (gene order doesn't matter)
predictions_match = np.allclose(y_pred_prob, y_pred_prob_shuffled)
print(f"Predictions identical with shuffled genes: {predictions_match}")
Testing gene-order independence...
2025-10-10 11:30:47 [info ] Downloading registry model 'pan_cancer_logistic' to cache directory: c:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\docs\notebooks\.scxpand_cache [scxpand.pretrained.download_manager]
2025-10-10 11:30:47 [info ] Loaded data format from: C:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\docs\notebooks\.scxpand_cache\5be9d6a5c323e8814ceae709733c7003-1.unzip\logistic\data_format.json [scxpand.data_util.data_format]
2025-10-10 11:30:47 [info ] Loading logistic model from C:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\docs\notebooks\.scxpand_cache\5be9d6a5c323e8814ceae709733c7003-1.unzip\logistic [scxpand.util.inference_utils]
2025-10-10 11:30:47 [info ] Inference environment ready: logistic model on cuda [scxpand.util.inference_utils]
2025-10-10 11:30:47 [info ] Running inference... [scxpand.core.prediction]
2025-10-10 11:30:47 [info ] Data transformation initialized: 11950 overlapping genes, 0 missing genes, 0 extra genes [scxpand.data_util.dataloaders]
2025-10-10 11:30:47 [info ] Created eval data loader with batch size: 2048, num_workers: 4 [scxpand.data_util.dataloaders]
2025-10-10 11:31:01 [info ] Inference completed. Generated 26596 predictions. [scxpand.core.prediction]
2025-10-10 11:31:01 [info ] Missing ['expansion'] columns in observation data. Skipping metrics evaluation. [scxpand.core.evaluation]
Inference with shuffled genes completed!
Example predictions (probabilities): [0.76589788 0.54469571 0.14568532 0.78571869 0.60704418]
Predictions identical with shuffled genes: True
Missing Gene Handling Test#
The model automatically handles missing genes by setting their expression to zero,
mimicking sequencing dropouts. Let’s test this by removing some genes.
# Remove last 50 genes to demonstrate missing gene handling
print("Testing missing gene handling...")
n_genes_to_remove = 50
adata_subset = adata_demo[:, :-n_genes_to_remove]
print(f"Removed {n_genes_to_remove} genes: {adata_subset.n_vars} genes remaining")
results = run_inference(
model_name=model_name,
adata=adata_subset,
batch_size=batch_size,
num_workers=num_workers,
)
y_pred_prob_missing = results.predictions
print("Inference with missing genes completed!")
print(f"Example predictions (probabilities): {y_pred_prob_missing[:5]}")
# Compare predictions with and without missing genes
print("\nPrediction comparison:")
print(f"Original predictions mean: {y_pred_prob.mean():.4f}")
print(f"Missing genes predictions mean: {y_pred_prob_missing.mean():.4f}")
print("Model successfully handled missing genes!")
Testing missing gene handling...
Removed 50 genes: 11900 genes remaining
2025-10-10 11:31:01 [info ] Downloading registry model 'pan_cancer_logistic' to cache directory: c:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\docs\notebooks\.scxpand_cache [scxpand.pretrained.download_manager]
2025-10-10 11:31:01 [info ] Loaded data format from: C:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\docs\notebooks\.scxpand_cache\5be9d6a5c323e8814ceae709733c7003-1.unzip\logistic\data_format.json [scxpand.data_util.data_format]
2025-10-10 11:31:01 [info ] Loading logistic model from C:\Users\KerenYlab\Documents\yizhak_ccg_scxpand\scXpand\docs\notebooks\.scxpand_cache\5be9d6a5c323e8814ceae709733c7003-1.unzip\logistic [scxpand.util.inference_utils]
2025-10-10 11:31:01 [info ] Inference environment ready: logistic model on cuda [scxpand.util.inference_utils]
2025-10-10 11:31:01 [info ] Running inference... [scxpand.core.prediction]
2025-10-10 11:31:01 [info ] Data transformation initialized: 11900 overlapping genes, 50 missing genes, 0 extra genes [scxpand.data_util.dataloaders]
2025-10-10 11:31:01 [info ] Created eval data loader with batch size: 2048, num_workers: 4 [scxpand.data_util.dataloaders]
2025-10-10 11:31:15 [info ] Inference completed. Generated 26596 predictions. [scxpand.core.prediction]
2025-10-10 11:31:15 [info ] Missing ['expansion'] columns in observation data. Skipping metrics evaluation. [scxpand.core.evaluation]
Inference with missing genes completed!
Example predictions (probabilities): [0.7643472 0.53746609 0.14428972 0.78419805 0.60384738]
Prediction comparison:
Original predictions mean: 0.5633
Missing genes predictions mean: 0.5648
Model successfully handled missing genes!
Summary#
This tutorial demonstrated:
Data Loading: How to load and combine multiple samples
Quality Control: Mitochondrial gene filtering and doublet detection
Data Preparation: T cell filtering and gene ID conversion
Model Application: Running inference with pretrained scXpand models
Robustness Testing: Gene order independence and missing gene handling
The scXpand model is now ready to predict T cell expansion probabilities on your data!