Model Inference & Evaluation Pipeline#
This script demonstrates how to:
Load trained models of different types
Run inference on new data using the loaded model.
Compute and plot the overall ROC curve (AUROC) using the data labels.
Compute and plot ROC curves per strata, where each strata is defined as the composite of
tissue_typeandimputed_labels.
Imports and Setup#
import sys
from pathlib import Path
import anndata as ad
import numpy as np
import seaborn as sns
# Set matplotlib backend for Jupyter notebooks
%matplotlib inline
project_root = Path.cwd().parent.parent
print(f"Project root: {project_root}")
sys.path.insert(0, str(project_root))
import scxpand
from scxpand.data_util.data_splitter import get_patient_identifiers
from scxpand.data_util.transforms import extract_is_expanded
from scxpand.util.general_util import metrics_dict_to_dataframes
from scxpand.util.plots import plot_roc_curve, plot_roc_curves_per_strata
sns.set_style("whitegrid")
Project root: c:\Users\Ron\repos\scXpand
Configuration#
Model configuration#
Local Model: Set
RESULTS_PATHto use a model you trained locally (default)Registry Model: Set
MODEL_NAMEto use a curated pre-trained model from scXpandDirect URL: Set
MODEL_URLto use any external model via direct URL
# Set ONE of the following inference modes:
# === OPTION 1: load model from local path
# RESULTS_PATH = project_root / "results/mlp"
# MODEL_NAME = None
# MODEL_URL = None
# === OPTION 2: load model from registry using the model name (choose from list_pretrained_models())
RESULTS_PATH = None
MODEL_NAME = "pan_cancer_autoencoder"
MODEL_URL = None
# === OPTION 3: load model from URL
# RESULTS_PATH = None
# MODEL_NAME = None
# MODEL_URL = "https://your-platform.com/model.zip"
Data input configuration#
# Choose ONE of the following options:
# Option 1: File-based inference (memory efficient for large datasets)
DATA_PATH = (
project_root / "data" / "scXpand_counts_with_expansion_for_model_08_12_2024.h5ad"
)
# Option 2: In-memory inference (faster for smaller datasets)
# Uncomment and modify one of these lines, then set DATA_PATH = None:
# adata = ad.read_h5ad("your_data.h5ad") # Load from file into memory
# adata = your_existing_adata_object # Use existing AnnData object
adata = None # Set this to your AnnData object for in-memory mode
# If using in-memory mode, set DATA_PATH = None
# DATA_PATH = None
# Optional: Use subset of data for evaluation
# If SPLIT_PATH is not None, only the patient IDs in the subset will be used for evaluation
SPLIT_PATH = project_root / "results" / "optuna_studies" / "dev_patient_ids.csv"
# Otherwise, the full dataset will be used for evaluation:
# SPLIT_PATH = None # Use full dataset
Additional configuration#
# Inference parameters
BATCH_SIZE = 2048
NUM_WORKERS = 4
# Set save path (if None, results will not be saved)
SAVE_PATH = None
# SAVE_PATH = project_root / "results/inference_results"
if SAVE_PATH:
SAVE_PATH.mkdir(parents=True, exist_ok=True)
Load Data#
The notebook automatically detects your data input mode:
File mode: If
DATA_PATHis provided andadatais NoneMemory mode: If
adatais provided andDATA_PATHis None
# Load data based on configuration
if adata is None and DATA_PATH is not None:
# File-based mode: load as backed for memory efficiency
print(f"Loading data from file: {DATA_PATH}")
adata = ad.read_h5ad(DATA_PATH, backed="r")
print(f"Loaded {adata.n_obs} cells, {adata.n_vars} genes (file-backed)")
elif adata is not None:
# Memory mode: adata already provided in configuration
print(f"Using provided AnnData object: {adata.n_obs} cells, {adata.n_vars} genes")
else:
raise ValueError("Must provide either DATA_PATH or adata in configuration")
Loading data from file: c:\Users\Ron\repos\scXpand\data\scXpand_counts_with_expansion_for_model_08_12_2024.h5ad
Loaded 1588309 cells, 11950 genes (file-backed)
# Get subset of data for evaluation (if specified)
if SPLIT_PATH:
with open(SPLIT_PATH) as f:
dev_patient_ids = [line.strip() for line in f]
patient_identifiers = get_patient_identifiers(obs_df=adata.obs)
eval_row_inds = np.where(patient_identifiers.isin(dev_patient_ids))[0]
else:
eval_row_inds = np.arange(len(adata))
n_cells_eval = len(eval_row_inds)
assert n_cells_eval > 0, "No cells found for evaluation"
print(
f"Evaluating on {n_cells_eval} cells ({n_cells_eval / len(adata) * 100:.2f}% of total)"
)
Evaluating on 331065 cells (20.84% of total)
# Run inference using the unified scXpand API
# Use the unified inference function - automatically handles all model types!
results = scxpand.run_inference(
data_path=DATA_PATH,
adata=adata,
model_path=RESULTS_PATH,
model_name=MODEL_NAME,
model_url=MODEL_URL,
save_path=SAVE_PATH,
batch_size=BATCH_SIZE,
num_workers=NUM_WORKERS,
eval_row_inds=eval_row_inds,
)
# Extract results
y_pred_prob = results.predictions
print(f"Generated predictions for {len(y_pred_prob)} cells")
print(f"Example predictions: {y_pred_prob[:5]}")
2025-09-17 10:39:59 [info ] Downloading registry model 'pan_cancer_autoencoder' to cache directory: c:\Users\Ron\repos\scXpand\docs\notebooks\.scxpand_cache [scxpand.pretrained.download_manager]
2025-09-17 10:40:00 [info ] Loaded data format from: C:\Users\Ron\repos\scXpand\docs\notebooks\.scxpand_cache\f876361c9c71e3c48397566cb0567d10-1.unzip\autoencoder\data_format.json [scxpand.data_util.data_format]
2025-09-17 10:40:00 [info ] Loading autoencoder model from C:\Users\Ron\repos\scXpand\docs\notebooks\.scxpand_cache\f876361c9c71e3c48397566cb0567d10-1.unzip\autoencoder [scxpand.util.inference_utils]
2025-09-17 10:40:00 [info ] Loaded data format from: C:\Users\Ron\repos\scXpand\docs\notebooks\.scxpand_cache\f876361c9c71e3c48397566cb0567d10-1.unzip\autoencoder\data_format.json [scxpand.data_util.data_format]
2025-09-17 10:40:00 [info ] Model input dimensions: genes=11950 [scxpand.autoencoders.ae_models]
2025-09-17 10:40:00 [info ] Building fork model with latent_dim=32 [scxpand.autoencoders.ae_models]
2025-09-17 10:40:00 [info ] Loading Autoencoder model from C:\Users\Ron\repos\scXpand\docs\notebooks\.scxpand_cache\f876361c9c71e3c48397566cb0567d10-1.unzip\autoencoder [scxpand.util.model_loading]
2025-09-17 10:40:01 [info ] Inference environment ready: autoencoder model on cuda [scxpand.util.inference_utils]
2025-09-17 10:40:01 [info ] Running inference... [scxpand.core.prediction]
2025-09-17 10:40:01 [info ] Created eval data loader with batch size: 2048, num_workers: 4 [scxpand.data_util.dataloaders]
2025-09-17 10:40:23 [info ] Inference: 12.3% | Time: 22.1s / ~179.1s [scxpand.util.general_util]
2025-09-17 10:40:33 [info ] Inference: 24.7% | Time: 32.7s / ~132.3s [scxpand.util.general_util]
2025-09-17 10:40:44 [info ] Inference: 37.0% | Time: 43.6s / ~117.6s [scxpand.util.general_util]
2025-09-17 10:40:57 [info ] Inference: 49.4% | Time: 56.4s / ~114.1s [scxpand.util.general_util]
2025-09-17 10:41:10 [info ] Inference: 61.7% | Time: 69.4s / ~112.4s [scxpand.util.general_util]
2025-09-17 10:41:22 [info ] Inference: 74.1% | Time: 81.0s / ~109.4s [scxpand.util.general_util]
2025-09-17 10:41:34 [info ] Inference: 86.4% | Time: 93.0s / ~107.6s [scxpand.util.general_util]
2025-09-17 10:41:46 [info ] Inference: 98.8% | Time: 104.8s / ~106.2s [scxpand.util.general_util]
2025-09-17 10:41:47 [info ] Inference completed. Generated 331065 predictions. [scxpand.core.prediction]
2025-09-17 10:41:48 [info ]
Evaluation Results (prediction)
===============================
Overall Metrics:
---------------
Metric Value
------------------- -------
AUROC 0.9238
F1 0.7496
RMSE 0.3302
error_rate 0.1531
false_negative_rate 0.124
false_positive_rate 0.1634
positives_rate 0.2616
AUROC F1 RMSE error_rate false_negative_rate false_positive_rate positives_rate
---------------------- -------- ------ ------ ------------ --------------------- --------------------- ----------------
is_CD4__Blood 0.975 0.7719 0.0984 0.0114 0.1789 0.0074 0.0234
is_CD8__Blood 0.9116 0.8613 0.351 0.166 0.0378 0.3138 0.5355
Double_Positive__Blood 0.9274 0.7627 0.3419 0.1469 0.0307 0.1843 0.2435
Double_Negative__Blood 0.9097 0.1628 0.2494 0.0877 0.4167 0.0828 0.0146
is_Treg__Blood nan 0 0.0309 0.0003 nan 0.0003 0
is_CD4__Tumor 0.875 0.4598 0.2595 0.0951 0.53 0.0541 0.0861
Double_Positive__Tumor 0.792 0.2736 0.3772 0.2149 0.3592 0.2051 0.0631
is_CD8__Tumor 0.7771 0.7706 0.4559 0.2999 0.0647 0.5745 0.5387
is_Treg__Tumor 0.7779 0.1762 0.2703 0.0892 0.8655 0.0299 0.0709
Double_Negative__Tumor 0.8972 0.5852 0.2936 0.1195 0.4203 0.0683 0.1454
average 0.8714 0.4824 0.2728 0.1231 0.3226 0.1521 0.1721
harmonic_avg 0.866 0 0.1467 0.0029 0.0992 0.0028 0
[scxpand.util.metrics]
2025-09-17 10:41:48 [info ] Metrics evaluation completed for prediction. AUROC: 0.9238 [scxpand.core.evaluation]
Generated predictions for 331065 cells
Example predictions: [0.02010737 0.488909 0.02501962 0.0180994 0.12366898]
Evaluate Predictions#
if results.has_metrics:
overall_df, category_df = metrics_dict_to_dataframes(results.metrics, precision=4)
# Display overall metrics
if overall_df is not None:
print("Overall Metrics:")
display(overall_df)
# Display category-specific metrics
if category_df is not None:
print("\nCategory-Specific Metrics:")
display(category_df)
Overall Metrics:
Category-Specific Metrics:
| Metric | Value | |
|---|---|---|
| 0 | AUROC | 0.9238 |
| 1 | F1 | 0.7496 |
| 2 | RMSE | 0.3302 |
| 3 | error_rate | 0.1531 |
| 4 | false_negative_rate | 0.1240 |
| 5 | false_positive_rate | 0.1634 |
| 6 | positives_rate | 0.2616 |
| AUROC | F1 | RMSE | error_rate | false_negative_rate | false_positive_rate | positives_rate | |
|---|---|---|---|---|---|---|---|
| is_CD4__Blood | 0.9750 | 0.7719 | 0.0984 | 0.0114 | 0.1789 | 0.0074 | 0.0234 |
| is_CD8__Blood | 0.9116 | 0.8613 | 0.3510 | 0.1660 | 0.0378 | 0.3138 | 0.5355 |
| Double_Positive__Blood | 0.9274 | 0.7627 | 0.3419 | 0.1469 | 0.0307 | 0.1843 | 0.2435 |
| Double_Negative__Blood | 0.9097 | 0.1628 | 0.2494 | 0.0877 | 0.4167 | 0.0828 | 0.0146 |
| is_Treg__Blood | nan | 0.0000 | 0.0309 | 0.0003 | nan | 0.0003 | 0.0000 |
| is_CD4__Tumor | 0.8750 | 0.4598 | 0.2595 | 0.0951 | 0.5300 | 0.0541 | 0.0861 |
| Double_Positive__Tumor | 0.7920 | 0.2736 | 0.3772 | 0.2149 | 0.3592 | 0.2051 | 0.0631 |
| is_CD8__Tumor | 0.7771 | 0.7706 | 0.4559 | 0.2999 | 0.0647 | 0.5745 | 0.5387 |
| is_Treg__Tumor | 0.7779 | 0.1762 | 0.2703 | 0.0892 | 0.8655 | 0.0299 | 0.0709 |
| Double_Negative__Tumor | 0.8972 | 0.5852 | 0.2936 | 0.1195 | 0.4203 | 0.0683 | 0.1454 |
| average | 0.8714 | 0.4824 | 0.2728 | 0.1231 | 0.3226 | 0.1521 | 0.1721 |
| harmonic_avg | 0.8660 | 0.0000 | 0.1467 | 0.0029 | 0.0992 | 0.0028 | 0.0000 |
Overall ROC Curve#
if results.has_metrics:
eval_obs = adata[eval_row_inds].obs
y_true = extract_is_expanded(eval_obs)
overall_auroc = plot_roc_curve(
labels=y_true,
probs_pred=y_pred_prob,
show_plot=True,
plot_save_dir=SAVE_PATH,
plot_name="roc_curve_overall",
title="ROC Curve (all cells)",
)
print(f"Overall AUROC (all cells): {overall_auroc:.5f}. Plot saved to {SAVE_PATH}")
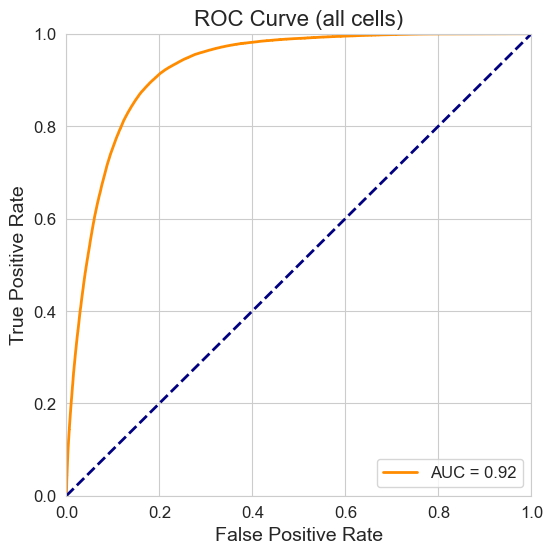
Overall AUROC (all cells): 0.92384. Plot saved to None
Per-Strata AUROC Evaluation#
# Per-strata evaluation
if results.has_metrics:
strata_cols = ["tissue_type", "imputed_labels"]
strata_df = eval_obs[strata_cols]
strata = strata_df.astype(str).agg(" - ".join, axis=1)
strata.index = eval_obs.index
unique_strata = strata.unique()
print(f"Unique strata found: {unique_strata}")
print(f"Harmonic Average AUROC per Strata: {results.get_harmonic_avg_auroc():.5f}")
Unique strata found: ['Blood - is_CD4' 'Blood - is_CD8' 'Blood - Double_Positive'
'Blood - Double_Negative' 'Blood - is_Treg' 'Tumor - is_CD4'
'Tumor - Double_Positive' 'Tumor - is_CD8' 'Tumor - is_Treg'
'Tumor - Double_Negative']
Harmonic Average AUROC per Strata: 0.86601
if results.has_metrics:
auroc_per_strata = plot_roc_curves_per_strata(
y_true=y_true,
y_pred_prob=y_pred_prob,
obs_df=eval_obs,
strata_columns=strata_cols,
show_plot=True,
plot_save_dir=SAVE_PATH,
max_cols=2,
)
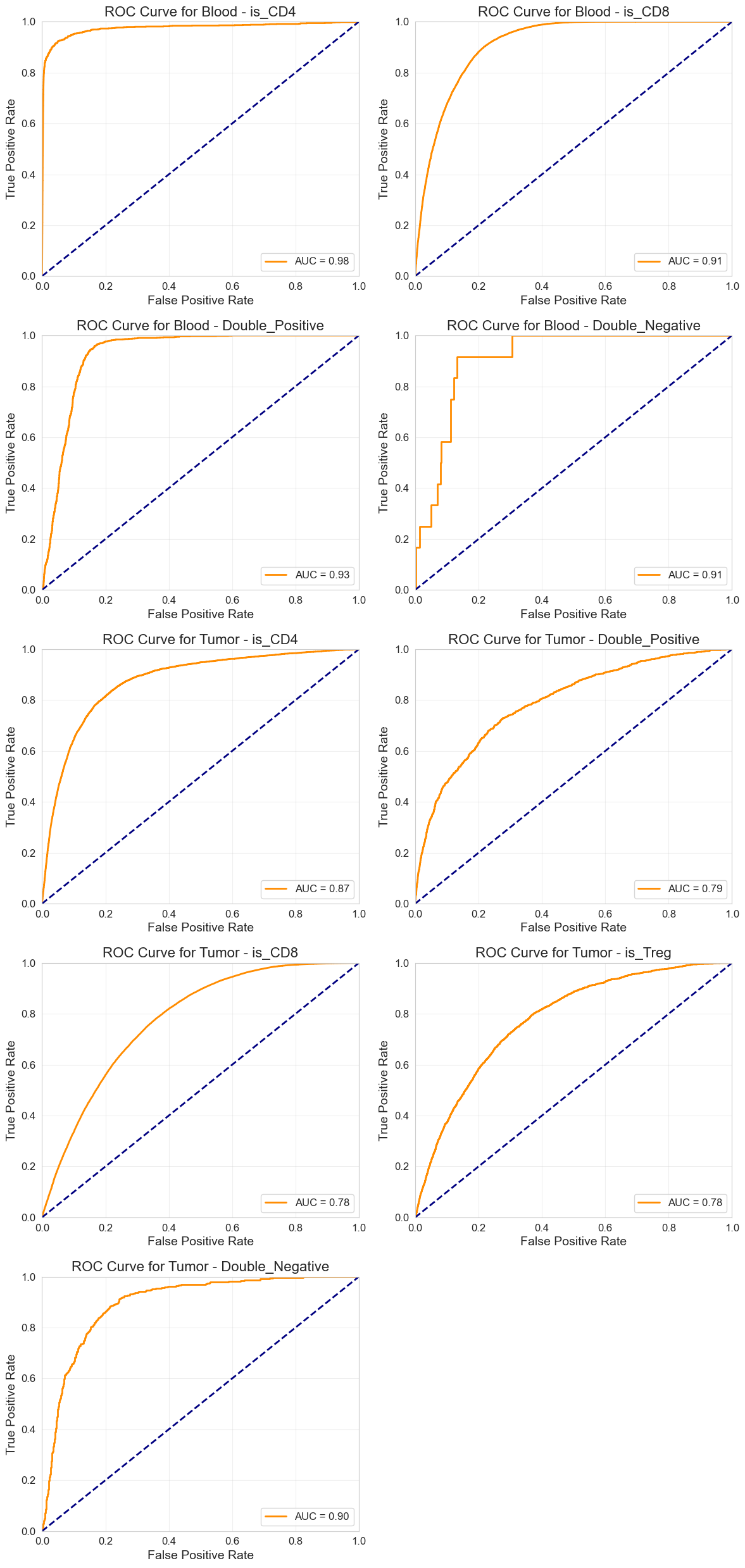
Save Predictions#
import pandas as pd
def save_predictions(
predictions: np.ndarray,
adata: ad.AnnData,
save_path: Path,
) -> None:
if not SAVE_PATH:
return
pred_column_name = "expansion_probability"
predictions_df = pd.DataFrame(
{"cell_id": adata.obs.index, pred_column_name: predictions}
)
csv_path = save_path / "predictions.csv"
try:
predictions_df.to_csv(csv_path, index=False)
print(f"Saved predictions to {csv_path}")
except Exception:
print(f"Failed to save predictions to {csv_path}")
raise
# Note: We need to create a subset of adata that matches the predictions
# Since predictions are already subset to eval_row_inds, we need matching adata subset
eval_adata_subset = adata[eval_row_inds]
save_predictions(predictions=y_pred_prob, adata=eval_adata_subset, save_path=SAVE_PATH)
# Clean up: close file if it was opened in backed mode
if hasattr(adata, "isbacked") and adata.isbacked:
adata.file.close()
print("Closed backed AnnData file")
Closed backed AnnData file